Пупочная грыжа или омфалоцеле

Омфалоцеле – врожденная аномалия передней брюшной стенки, при которой органы брюшной полости выходят за ее пределы в составе грыжевого мешка. Клинически патология проявляется эвентрацией петель кишечника, желудка, печени и других органов, прикрытых висцеральной брюшиной, через грыжевые ворота в участке пупочного кольца. Антенатальная диагностика включает в себя УЗИ ОБП, определение уровня α-фетопротеина, амниоцентез с дальнейшим кариотипированием. Лечение хирургическое, предусматривает радикальное или поэтапное погружение содержимого грыжевого пешка обратно в брюшную полость с последующей пластикой передней брюшной стенки.
Общие сведения
Омфалоцеле (эмбриональная грыжа, грыжа пупочного канатика) – аномалия развития, при которой происходит выпячивание органов брюшной полости, укрытых висцеральной брюшиной, сквозь переднюю брюшную стенку. Впервые данную патологию описал французский хирург А. Паре в 1634 году. В среднем заболеваемость составляет порядка 2,2:10000 новорожденных. Точную частоту, с которой встречается омфалоцеле, установить невозможно, поскольку в большинстве случаев такие беременности прерываются. У мальчиков данная патология возникает в 1,5 раза чаще, чем у девочек. Наибольшая склонность наблюдается у представителей европеоидной расы – 85% от всех случаев омфалоцеле. Для представителей негроидной расы этот показатель составляет 13%, монголоидной – 2%. Примерно 55% случаев патологии диагностируется при беременности у женщин в возрасте старше 35 лет. Общая летальность зависит от сопутствующих расстройств и колеблется от 9 до 60% новорожденных. При изолированной форме и адекватном лечении прогноз для жизни и здоровья ребенка благоприятный.
Омфалоцеле
Причины омфалоцеле
Омфалоцеле – это гетерогенное заболевание, при котором нарушается процесс внутриутробного вправления физиологической пупочной грыжи. Патогенетически это может быть результатом пороков развития пупочного кольца и передней брюшной стенки, генетических аномалий, неполного погружения органов обратно в брюшную полость или дефектов строения кишечника.
При внутриутробном развитии кишечника плода, а именно – в процессе его трансформации от первичной кишечной трубки до зрелых петель происходит его физиологической разворот. Данный процесс начинается с 5 недели беременности. От этого момента и до 10 недели кишечник сильно увеличивается в объеме, из-за чего не помешается в брюшной полости. Петли давят на брюшную стенку, формируя физиологическую пупочную грыжу. К концу 10 недели абдоминальная полость стремительно прибавляет в объеме, на фоне чего происходит самостоятельное вправление эвентрации. Если данный механизм нарушается силу различных патологических изменений, возникает омфалоцеле.
Способствовать возникновению данной патологии могут вредные привычки матери (алкоголь, курение, наркотики), нерациональный прием медикаментов, беременность после 35 лет, при которой растет риск хромосомных аномалий – синдромов Патау, Эдвардса. Довольно часто омфалоцеле выступает в роли одного из симптомов таких патологий, как синдром Беквита-Видемана, пентада Кантрелла, синдром амниотических тяжей, порок развития стебля тела, OEIS комплекса. Крайне редко удается установить наследственную склонность, что свидетельствует о возможной генетической предрасположенности.
Классификация и симптомы омфалоцеле
В педиатрии в зависимости от размера дефекта и его содержания выделяют следующие формы омфалоцеле:
- Малая. Диаметр – до 5 см. Наиболее распространенная форма. Содержит 1-2 кишечные петли. В основном выступает в роли проявлений хромосомных аномалий.
- Средняя. Размер дефекта пупочного кольца – от 5 до 10 см. В составе грыжевого мешка содержатся 2-4 кишечные петли.
- Большая. Дефект передней брюшной стенки составляет более 10 см. Помимо петель кишечника через него выходит часть печени, желудок и другие органы.
По наличию сопутствующих патологий выделяют:
- Изолированное омфалоцеле. Грыжа пупочного канатика – единственная развившаяся внутриутробная патология.
- Сочетанная форма. Помимо дефекта пупочного кольца у ребенка присутствуют хромосомные мутации (25-35%), пороки развития сердечно-сосудистой (15-50%) и мочеполовой систем (до 15%). Характерны грыжи пищеводного отверстия диафрагмы и дисплазии тазобедренных суставов, другие скелетные аномалии.
Омфалоцеле является врожденной патологией, которая диагностируется еще в антенатальном периоде. После родов общее состояние ребенка зависит от сопутствующих заболеваний и срока гестации. При омфалоцеле характерны преждевременные роды, масса тела ребенка 1500г и меньше. Объективные признаки визуализируются уже с момента рождения. Определяется дефект передней брюшной стенки, который локализирован по срединной линии на уровне пупочного кольца. В этом месте находится образование, представленное петлями тонкого и толстого кишечника, возможно – желудком и печенью, покрытыми висцеральными листком брюшины. В 10-20% наблюдается анте- или интранатальный разрыв грыжевого мешка. Пуповина также входит в состав омфалоцеле. Абдоминальные мышцы развиты нормально. Другие возможные пороки развития зависят от сопутствующих патологий и могут включать в себя деформации позвоночного столба и конечностей, макроглоссию, макросомию, атрезию ануса и т. д.
Диагностика омфалоцеле
В современных условиях наличие омфалоцеле определяется еще в I триместре беременности благодаря инструментальным и лабораторным методам исследования. Ведущую роль играет УЗ-диагностика. В зависимости от аппарата установить наличие данной патологии можно уже на 11-14 неделе. Основной признак – эвентрация петель кишечника, печени, желудка за пределы брюшной полости в участке крепления пуповины к брюшной стенке. Все вышедшие органы покрыты мембраной, состоящей из брюшины, вартонового студня и амниотической оболочки. Наличие данной мембраны важно для проведения дифференциальной диагностики с гастрошизисом. При внутриутробном разрыве грыжевого мешка петли кишечника прикрывает только амнион. Также на потенциальное развитие омфалоцеле может указывать размер ворот физиологической грыжи, составляющий свыше 7 мм в диаметре. УЗИ позволяет выявить другие проявления присутствующих генетических патологий. Повторные исследования проводятся каждые 2-3 недели, т. к. возможно самостоятельно закрытие дефекта в более поздние сроки.
Лабораторная диагностика омфалоцеле осуществляется при помощи биохимического исследования крови с измерением уровня α-фетопротеина. При врожденных пороках развития этот показатель будет значительно выше нормы для имеющегося срока беременности. Для подтверждения генетических аномалий показано проведение амниоцентеза с дальнейшим кариотипированием.
Лечение омфалоцеле
При антенатальной постановке диагноза омфалоцеле роды проводятся в специализированном перинатальном центре в условиях развернутой операционной. При малых и средних размерах дефекта пупочного кольца и отсутствии угрожающих жизни матери и ребенка состояний родоразрешение может осуществляться через естественные родовые пути. Во всех других ситуациях показано кесарево сечение в связи с большим риском интранатального разрыва грыжевого мешка. Дальнейшая терапевтическая тактика зависит общего состояния ребенка, размеров пупочной грыжи и сопутствующих патологий.
Консервативная терапия используется при невозможности провести хирургическое вмешательство. Как правило, она применяется при больших формах омфалоцеле и комбинации с множественными тяжелыми аномалиями развития. Лечение заключается в формировании плотной корки и рубца, что трансформирует данную патологию в массивную вентральную грыжу, которую в дальнейшем оперируют. С этой целью применяют дубящие средства (5% перманганат калия, нитрат серебра), которые наносят на вертикально зафиксированный за пуповину грыжевой мешок 2-3 раза в день. Консервативное лечение используется крайне редко, т. к. при нем имеется большая вероятность инфицирования и сепсиса, риск разрыва оболочек, массивной спаечной болезни в дальнейшем. Предпочтение отдается раннему оперативному вмешательству.
Хирургическая тактика при омфалоцеле напрямую зависит от размеров дефекта брюшной стенки, операции могут проводиться в один или несколько этапов. Начинают такое лечение на протяжении первых 1-2 дней жизни ребенка. При одномоментном вмешательстве выполняется погружение содержимого грыжевого мешка в абдоминальную полость и послойное зашивание брюшной стенки с ее пластикой и формированием пупка. Такая тактика – метод выбора, при омфалоцеле малых и средних размеров с минимальной торакоабдоминальной диспропорцией. В других случаях прибегают к поэтапному лечению. Первый этап – подшивание силиконового мешка с силопластиковым покрытием и помещением в него содержимого грыжевого мешка. По мере постепенного погружения органов в абдоминальную полость данный мешок перевязывают, уменьшая его в объеме. На 5-15 день выполняется второй этап – удаление мешка и формирование минимальной вентральной грыжи посредством ушивания дефекта. В возрасте 5-7 месяцев эта грыжа удаляется, после чего осуществляется полноценная пластика брюшной стенки.
После первого этапа или одноэтапной операции ребенок помещается в кувез для поддержания температурного режима, применяются обезболивающие, антибиотики широкого спектра действия. Режим питания парентеральный. При необходимости выполняется декомпрессия желудка через назо- или орогастральный зонд, проводится инфузионная терапия и ИВЛ. После проведенного лечения следует длительный реабилитационный период, в течение которого осуществляется коррекция всех послеоперационных осложнений.
Прогноз и профилактика омфалоцеле
В целом прогноз для детей с омфалоцеле сомнительный. Исход зависит от срока гестации, сопутствующих хромосомных аномалий и анатомических пороков развития, диаметра дефекта пупочного кольца, объема и содержания грыжевого мешка, результатов проведенного лечения. Как правило, при адекватной терапии и отсутствии сопутствующих патологий, несовместимых с жизнью, вероятность благоприятного исхода достаточно велика. При изолированной форме омфалоцеле малого размера, несмотря возможные отдаленные осложнения хирургического лечения (ГЭРБ, спаечная болезнь, пупочная грыжа), дальнейший рост и развитие ребенка не будут отличаться от возрастной нормы. На протяжении всего периода реабилитации показано диспансерное наблюдение у лечащего педиатра, хирурга и семейного врача.
Профилактика заключается в медико-генетическом консультировании семейных пар, планировании беременности, полном отказе матери от вредных привычек. С целью ранней диагностики омфалоцеле необходимо регулярно посещать женскую консультацию и проходить соответствующие обследования: УЗИ и измерение α-фетопротеина крови.
Источник
В процессе развития эмбриона, между 6-й и 10-й неделями, происходит увеличение размеров кишечника, его удлинение, и петли кишечника, не помещаясь в брюшной полости, выталкиваются за её пределы через пупочное кольцо в месте прикрепления пуповины к передней брюшной стенке. Располагаясь внебрюшинно, в пуповинных оболочках, они проходят временную стадию «физиологической кишечной грыжи», а затем, выполнив процесс вращения, возвращаются в увеличивающуюся брюшную полость. Если в результате нарушения процесса вращения кишечника, недоразвития брюшной полости или нарушения замыкания брюшной стенки часть органов остается в пуповинных оболочках, ребенок рождается с грыжей пупочного канатика, или ОМФАЛОЦЕЛЕ.К 11-й неделе беременности, в норме, петли кишечника возвращаются обратно в брюшную полость и грыжевое выпячивание исчезает.
К тому времени, когда вы приходите на скрининг 1-го триместра в 11-13 недель, в большинстве случаев этот процесс уже завершается, но следует помнить, что до 12-13 недель кишечник эмбриона может в норме выходить за границу брюшной полости, и это всё ещё будет считаться физиологической кишечной грыжей. Выпячивание петель кишечника в пуповину, происходящее при нормальном развитии, обычно сопровождается увеличением диаметра ее основания менее чем на 7 мм.
В случае, когда вышедшее кишечное содержимое определяется отдельно от области вхождения пуповины в брюшную полость и не покрыто оболочкой, то такая эхографическая картина будет являться диагностическим признаком уже другой аномалии развития передней брюшной стенки – гастрошизиса, даже в первом триместре беременности.
При расчёте риска хромосомных аномалий (ХА) в программном обеспечении FMF в 11-13 недель, обнаружение омфалоцеле хоть и указывается, но на результат не влияет.
Итак. Омфалоцеле представляет собой дефект передней брюшной стенки в области пупочного кольца с образованием грыжевого мешка с внутрибрюшинным содержимым, покрытого амниоперитонеальной мембраной.
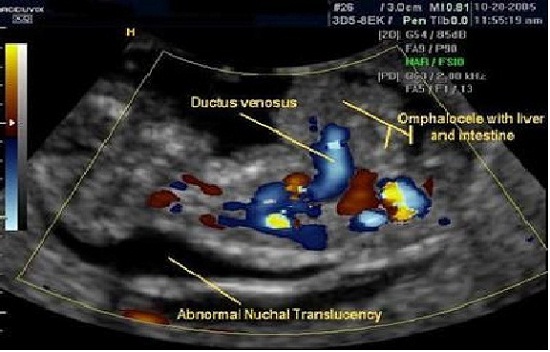
При рождении ребёнка омфалоцеле может быть маленьким, лишь с небольшой частью петли кишечника, но может быть и большим, до 10см и более, включать в себя, помимо кишечника, печень и другие органы. Частота встречаемости маленьких омфалоцеле (до 5 см) – 1:5000 живорождённых, больших (10 см и более) – 1:10000 живорождённых.
Ультразвуковая диагностика омфалоцеле основана на обнаружении примыкающего к передней брюшной стенке образования округлой или овальной формы с чёткими ровными контурами, заполненного неоднородным содержимым, непосредственно к которому прикрепляется пуповина.
Омфалоцеле может быть изолированным, когда никаких других изменений и аномалий развития у плода нет.
Однако:
- Примерно в 30% случаев отмечается сочетание омфалоцеле с хромосомными аномалиями. Согласно результатам Е.В. Юдиной, частота ХА при омфалоцеле у плода в группе пациенток старше 35 лет составила 54,5%, а у пациенток моложе 35 лет – 28%. Все хромосомные аномалии были обнаружены только в случаях сочетанных омфалоцеле, их частота в этой группе составила 46,4%;
- У части детей впоследствии диагностируется синдром Беквита-Видемана;
- Больше чем у половины детей с омфалоцеле обнаруживаются пороки других органов и систем. Чаще всего это пороки сердечно-сосудистой, мочеполовой системы, позвоночника, диафрагмальная грыжа, скелетные дисплазии.
Что делать, если во время скрининга первого триместра был поставлен диагноз омфалоцеле
- Помните, что до 12-13 недель это может быть вариантом нормы и дождитесь результатов скринингового обследования.
- Если по результатам скрининга индивидуальный риск ХА низкий, то повторите УЗИ через 2-3 недели. К этому времени физиологическая кишечная грыжа должна исчезнуть.
- При высоком риске ХА, в любом случае показано пренатальное медико-генетическое консультирование и рекомендовано проведение кариотипирования плода. Либо вы можете сделать выбор в пользу прерывания беременности.
- При повторном обнаружении омфалоцеле спустя 2-3 недели, даже при низком индивидуальном риске ХА в первом триместре, показано пренатальное медико-генетическое консультирование, рекомендовано проведение кариотипирования плода, а также тщательная оценка ультразвуковой анатомии плода в 18-20 недель для исключения сочетанных пороков развития.
При пролонгировании беременности, показано динамическое ультразвуковое наблюдение каждые 3-4 недели для оценки роста плода и динамики изменений размеров грыжевого мешка. В среднем в 25% случаев отмечается внутриутробная задержка роста плода, но следует учитывать, что точность фетометрии в этом случае невысока, так как окружность живота значительно изменена. Иногда происходит разрыв оболочек омфалоцеле, и тогда становится практически невозможно отличить грыжу пупочного канатика от гастрошизиса.
Методом выбора при родоразрешении часто является кесарево сечение в плановом порядке в интересах плода, хотя в этом вопросе мнения специалистов неоднозначны.
После рождения, при совсем маленьких грыжах (до 1.5 см), можно надеяться на самостоятельное закрытие пупочного кольца через несколько недель, но если по достижении трехлетнего возраста грыжа самостоятельно не закрылась, принимается решение об операции. Более крупные омфалоцеле, как правило, оперируют в первые сутки жизни. Большие размеры грыжевого мешка требуют двухэтапной операции с постепенным вправлением органов в брюшную полость.
Kaiser и соавторы проследили развитие детей на протяжении 1-28 лет, прооперированных по поводу омфалоцеле, и пришли к выводу, что наличие изолированного омфалоцеле не является показанием к прерыванию беременности, так как последующее развитие и социальная адаптация детей протекает без осложнений.
Несколько слов о синдроме Беквита-Видемана
Это генетически-обусловленное заболевание, которое характеризуется сочетанием макросомии (быстрый, гипертрофичный рост), омфалоцеле, макроглоссии (большой, не помещающийся во рту язык), предрасположенностью к эмбриональным опухолевым образованиям и неонатальной гипогликемией (снижение уровня сахара в крови у новорожденного). Кроме того, часто встречаются аномалии развития ушных раковин и гемигиперплазия, когда некоторые части тела на одной стороне больше, чем на другой.
Такие дети рождаются достаточно крупными (около 4 кг) и в первые месяцы жизни значительно прибавляют в весе и росте. При рождении обращает на себя внимание большой, высовывающийся изо рта язык и дефект передней брюшной стенки, в основном омфалоцеле.
[IMG ID=324]
При своевременной коррекции гипогликемии, ускоренные темпы роста постепенно снижаются уже в детском возрасте, интеллектуальное развитие, как правило не страдает, и взрослые люди обычно не испытывают медицинских проблем, связанных с этим заболеванием. Но ранняя диагностика синдрома Беквита-Видемана важна, так как у таких детей повышенный риск образования различных опухолей, в первую очередь это опухоль Вильмса (нефробластома) и гепатобластома. После 10 летнего возраста риск образования опухолей снижается до общепопуляционного.
Заподозрить синдрома Беквита-Видемана во время УЗИ возможно, но только в III триместре при сочетании макроглоссии, омфалоцеле и макросомии с нормальным кариотипом. В журнале Пренатальная диагностика за 2003 год было представлено описание случая пренатальной ультразвуковой диагностики синдрома Беквита – Видемана без омфалоцеле у плода в III триместре беременности. Подозрение о наличии указанного синдрома было высказано при ультразвуковом исследовании в 30-31 неделю беременности на основании макроглоссии, микроринии, висцеромегалии и макросомии. Диагноз подтвержден после рождения ребенка.
Источники:
1. https://medicalplanet.su/akusherstvo/174.html
2. https://omphalocele.net/
3. https://stopgryzha.ru/belly/fiziologicheskaya-embrionalnaya-gryizha-u-ploda-omfalotsele
4. https://www.cancer.net/cancer-types/beckwith-wiedemann-syndrome
Источник
Медицинский эксперт статьи
х
Весь контент iLive проверяется медицинскими экспертами, чтобы обеспечить максимально возможную точность и соответствие фактам.
У нас есть строгие правила по выбору источников информации и мы ссылаемся только на авторитетные сайты, академические исследовательские институты и, по возможности, доказанные медицинские исследования. Обратите внимание, что цифры в скобках ([1], [2] и т. д.) являются интерактивными ссылками на такие исследования.
Если вы считаете, что какой-либо из наших материалов является неточным, устаревшим или иным образом сомнительным, выберите его и нажмите Ctrl + Enter.
Омфалоцеле (грыжа пупочного канатика, пуповинная грыжа, эмбриональная грыжа) – это выпячивание органов брюшной полости через дефект средней линии в основании пупка.
Омфалоцеле – аномалия развития, при которой в результате раннего нарушения органогенеза органы брюшной полости в той или иной степени развиваются вне туловища эмбриона, что влечёт за собой не только неправильное развитие этих органов, но и дефекты формирования как брюшной полости, так и грудной клетки. Грыжевое выпячивание покрыто грыжевым мешком, состоящим снаружи от амниона, изнутри – из брюшины, с мезенхимой (Варгановым студнем) между ними.
При омфалоцеле выпячивание органов покрыто тонкой оболочкой и может быть маленьким (только несколько петель кишечника) или может содержать большую часть органов брюшной полости (кишечник, желудок, печень). Немедленная опасность заключается в высыхании внутренних органов, гипотермии и дегидратации из-за испарения воды, а также инфицирования брюшины. У новорожденных с омфалоцеле отмечается очень высокая частота других врожденных пороков развития, включая атрезию кишечника; хромосомные аномалии, например синдром Дауна; и аномалии сердца и почек, которые следует выявить и обследовать до хирургической коррекции.
Код по МКБ-10
Q79.2. Экзомфалоз.
Эпидемиология
Частота порока омфалоцеле составляет 1-2 на 10 000 живых новорождённых, особых различий по полу нет. Большая часть детей с омфалоцеле рождаются доношенными.
[1], [2], [3], [4], [5], [6], [7], [8], [9], [10]
Что вызывает омфалоцеле?
Представления об этологии и патогенезе омфалоцеле до сих пор остаются разноречивыми. Считают, что в генезе эмбриональной грыжи пупочного канатика основную роль играют два фактора – нарушение вращения кишечника в первой периоде поворота и недоразвитие передней брюшной стенки. Нарушение вращения кишечника проявляется в виде сохранения временной «физиологической» пупочной грыжи, образующейся у 5-недельного эмбриона из-за несоответствия темпов роста кишки и брюшной полости и самостоятельно исчезающей к 11-й нед развития.
Согласно другой теории, омфалоцеле представляет собой «персистирование туловищного стебелька в той области, которая в норме занята соматоплеврой». Это представление о нарушении латерального мезодермального замещения брюшины, амниона и мезодермы стебелька объясняет многообразие аномалий, отмечаемых при омфалоцеле – от клоакальной экстрофии до пентады Кантрелла.
Как проявляется омфалоцеле?
У детей с омфалоцеле часто встречают множественные сочетанные пороки других жизненно важных органов и систем, а также хромосомные аномалии. Чаше всего это врождённые пороки сердца, пороки развития почек, ортопедические пороки и др. Омфалоцеле малых размеров довольно часто сочетается с незаращенным желточным протоком.
Возможно сочетание омфалоцеле с болезнью Дауна, трисомией по хромосомам 13 и 18.
Омфалоцеле – компонент синдрома Беквита-Видеманна, носящего также наименование синдрома OMG (omphalocele-macroglossia-gigantism). Для этого синдрома, кроме омфалоцеле, характерны (как следует из названия) наличие большого языка, вызывающее иногда затруднение дыхания, и гигантизм, чаще всего реализованный в гигантизме паренхиматозных органов (гепатоспленомегалия, гиперплазия поджелудочной железы), что может проявляться гиперинсулинизмом и гипогликемией, особенно опасной в периоде новорождённости. Реже обнаруживают парциальный гигантизм скелета.
Омфалоцеле иногда бывает компонентом и таких тяжелейших аномалий, как пентада Кантрелла и клоакальная экстрофия, лечение которых представляет огромные трудности и до сих пор в большинстве клиник имеет неутешительные результаты. Именно тяжесть сочетанных поражений и их курабельность обусловливают тяжесть состояния бального с омфалоцеле и прогноз, а в танатогенезе или инвалидизации: пациента ведущая роль нередко принадлежит не омфалоцеле, а сочетанным порокам развития или генетическим синдромам. Всё вышесказанное диктует необходимость раннего выявления омфалоцеле в антенатальном периоде для своевремениого решения вопроса о сохранении или прерывании беременности.
Классификация
Согласно рабочей классификации омфалоцеле, в зависимости от размера дефекта передней брюшной стенки (грыжевых ворот) и объёма содержимого грыжевого мешка выделяют омфалоцеле малых, средних и больших размеров. Содержимым малых и средних грыж бывают тальке кишечные петли (в малой – одна или несколько). Большое омфалоцеле всегда содержит не только кишечные петли, но и печень.
По форме грыжевого выпячивания выделяют полушаровидные, шаровидные и грибовидные грыжи.
[11], [12], [13], [14], [15], [16], [17], [18], [19]
Как распознать омфалоцеле?
Визуализация омфалоцеле возможна при УЗИ с 14-й недели беременности. Весьма информативен тест на содержание у матери альфа-фетопротеина (АФП), его содержание повышено при врождённых пороках развития. В этом случае (при повышении количества АФП) необходимо тщательно обследовать плод на наличие сочетанных врождённых пороков развития. При выявлении омфалоцеле в сочетании с некурабельными пороками развития или генетическими аномалиями будущим родителям можно рекомендовать прерывание беременности.
Рождение детей с малым или средним омфалоцеле может происходить естественным путём, если нет других показаний для проведения кесарева сечения. При больших ГПК метод родоразрешения выбирают индивидуально в каждом конкретном случае. Обычно целесообразно выполнить кесарево сечение в связи с опасностью разрыва тонких оболочек грыжи.
Пренатальная диагностика омфалоцеле
Диагностика омфалоцеле после рождения ребёнка, как правило, не вызывает трудностей. Однако при омфалоцеле малых размеров при обработке пуповины в родильном доме могут быть допущены ошибки, имеющие тяжёлые последствия. Обычно в грыжевых оболочках при этом типе аномалии находится одна или две петли кишки, т.е. объём образования небольшой, и такое омфалоцеле нередко выглядит как утолщённая пуповина. Если врач или акушерка не распознали малое омфалоцеле и раздавливающую клемму или лигатуру наложили на границу между тенями пуповины и кожей, а остаток пуповины отсекли, то может быть повреждена стенка кишки. Поэтому в сомнительных случаях (при толстой пуповине, дисплазии сосудов пуповины) важно помнить об омфалоцеле малых размеров и накладывать лигатуру на расстоянии не менее 10-15 см от кожного края. Такому новорождённому нужен незамедлительный перевод в хирургический стационар для обследования. Подтвердить или исключить диагноз омфалоцеле малых размеров позволяет рентгенологическое обследование в боковой проекции. При омфалоцеле за пределами передней брюшной стенки в оболочках пуповины определяют кишечные петли (газовые пузыри), тогда как при отсутствии сообщения между брюшной полостью и оболочками пуповины целостность передней брюшной стенки на рентгенограмме не нарушена. Учитывая тот факт, что при омфалоцеле нередки сочетанные пороки развития, в обязательный протокол обследования больного, кроме рентгенографии органов грудной клетки и брюшной полости в вертикальном положении, входит ультрасонография головного мозга, брюшной полости и забрюшинного пространства, а также УЗИ сердца крупных сосудов.
Лечение омфалоцеле
При первой помощи ребёнку с омфалоцеле в родильном доме основное внимание следует уделять поддержанию температуры его тела, защите грыжевого мешка от неблагоприятных внешних воздействий. Больные с омфалоцеле нуждаются в экстренной помощи.
Выбор метода лечения омфалоцеле зависит от размеров грыжи, состояния больного и возможностей стационара, где проходит это лечение. Оно может быть консервативным или хирургическим и проходить в один или несколько этапов.
Консервативное лечение омфалоцеле
Консервативное лечение в последние годы по мере развития реаниматологии и улучшения реанимационной поддержки применяют в крайне ограниченных случаях, когда по тем или иным причинам предполагают отложить оперативное вмешательство. Подобную тактику можно использовать при огромных грыжах пупочного канатика или их сочетании с множественными тяжёлыми пороками развития. Наиболее часто для этих целей применяют такие дубящие растворы, как повидон-йод, мербромин, 5% раствор калия перманганата. Грыжевой мешок за пуповинный остаток фиксируют над больным в вертикальном положении, оболочки грыжи несколько раз в сутки обрабатывают одним из перечисленных растворов, добиваясь образования плотной корки, под которой постепенно формируется рубец, образуя большую вентральную грыжу. Однако этот метод имеет множество серьёзных недостатков (инфицирование оболочек, их разрыв, длительный период заживления, выраженный спаечный процесс и др.), поэтому его следует использовать лишь в экстраординарных случаях.
[20], [21], [22], [23], [24], [25], [26], [27], [28], [29], [30], [31]
Хирургическое лечение омфалоцеле
Хирургическое лечение может быть радикальным (послойное ушивание всех слоев брюшной стенки после погружения органов в брюшную полость) или этапным. Второй вариант предусматривает постепенное формирование передней брюшной стенки с использованием на промежуточных этапах ауто- или аллопластических материалов.
Радикальное вмешательство – операция выбора, выполняемая в тех случаях, когда висцеро-абдоминальная диспропорция (соотношение между объёмом грыжевого образования и ёмкостью брюшной полости) выражена умеренно, и послойное ушивание передней брюшной стенки не вызывает значительного повышения внутрибрюшного давления. Соответственно радикальную операцию обычно выполняют при омфалоцеле малых и средних размеров, реже – при больших омфалоцеле.
Если омфалоцеле малых размеров сочетается с желточным протоком, радикальное вмешательство дополняют резекцией желточного протока. Следует помнить, что компонентом практически любого омфалоцеле, за редким исключением при малых грыжах, бывает мальротация, общая брыжейка тонкой и толстой кишки, поэтому при погружении органов в брюшную полость толстая кишка должна отойти в левый фланг, а тонкая – находиться в правом фланге и центре брюшной полости. После завершения внутрибрюшинного этапа операции производят послойное ушивание раны передней брюшной стенки с формированием «косметического» пупка.
Наибольшие трудности возникают при лечении омфалоцеле больших размеров со значительной степенью висцеро-абдомннальной диспропорции, когда радикальная операция невозможна из-за резкого повышения внутрибрюшного давления. В таких случаях приходится применять различные виды этапного хирургического лечения.
В194S г. Роберт Гросс из Бостона описал метод этапного хирургического лечения больших грыж пупочного канатика. Первый этап заключался в удалении оболочек грыжи, погружении органов, насколько это возможно, в брюшную полость широкой отсепаровке кожных лоскутов брюшной стенки вплоть до поясничной области и ушивании кожи с формированием вентральной грыжи. Вторым этапом проводили ликвидацию вентральной грыжи (в возрасте L-2 лет). В настоящее время эту методику практически не используют, поскольку она имеет множество недостатков (выраженный спаечный процесс, большие размеры вентральной грыжи, отсутствие условий для увеличения объёма брюшной полости, так как почти все органы находятся в кожном грыжевом мешке).
Стремительный скачок в лечении больших омфалоцеле был сделан в 1967 г.. когда Шустер описал способ временного применения пластикового покрытия для уменьшения размеров фасциального дефекта.
3атем в 1969 г. Allen и Wrenn предложили использовать однослойное силастиковое покрытие, подшиваемое к краям фасциального дефекта с последующим постепенным уменьшением объёма грыжевого образования с помощью мануальной компрессии, что позволяет произвести отсроченное первичное закрытие борюшной стенки. Как только после первого этапа вмешательства восстановится моторика кишечника, он опорожнится и уменьшится в объёме, следует второй – (обычно через 3-14 дней) – удаление мешка и радикальная пластика передней брюшной стенки или формирование теперь уже небольшой вентральной грыжи. Этот метод остаётся главным в лечении данной патологии и в настоящее
Техника операции при этапном лечении больших омфалоцеле. Операцию начинают с разреза кожи вокруг грыжевого образования. Убедившись в невозможности погрузить все органы, к мышечно-апоневротическому краю дефекта передней брюшной стенки подшивают силиконовый мешок с силастиковым покрытием. Этим мешком покрывают ту часть содержимого грыжи, которую удалось поместить в брюшную полость. Мешок завязывают над органами, фиксируют над больным в вертикальном положении. По мере того как органы из мешка спонтанно опускаются в брюшную полость, мешок перевязывают всё ниже и ниже (по отношению к брюшной стенке), уменьшая его объём, при этом допускается некоторая степень компрессии. Второй этап заключается в удалении мешка через 7-14 дней) и радикальном послойном ушивании передней брюшной стенки в формировании небольшой вентральной грыжи. В этом случае последний этап оперативного лечения (ликвидация вентральной грыжи с послойным ушиванием брюшной стенки) производят в возрасте 6 мес.
Существуют и успешно используются методики применения аллотрансплантации синтетического или биологического происхождения, вшиваемых в фасциальный дефект передней брюшной стенки в виде заплаты при выраженной степени висцероабдоминальной диспропорции.
Послеоперационное ведение
В раннем послеоперационном периоде проводят искусственную вентиляцию легких, обезболивание, антибактериальную терапию. Решающий компонент лечения – тотальное парентеральное питание на протяжении всего периода заживления брюшной стенки и восстановления функций кишечника. У детей с сочетанными тяжёлыми аномалиями в послеоперационном периоде следует решить вопрос своевременной коррекции этих аномалий, что требует участия в лечении врачей этих специальностей. Особое внимание нужно уделять пациентам с синдромом Бесквита-Видеманна, склонным к тяжёлой гипогликемии. Тщательный контроль – держания сахара в крови позволяет предотвратить это состояние и не допустить развития энцефалопатии у таких больных.
Прогноз омфалоцеле
Все пациенты с омфалоцеле, не имеющие летальных пороков развития других органов и систем, выживают. Однако при сочетании омфалоцеле с различными аномалиями своевременная их диагностика, а также сотрудничество с врачами других специальностей позволяют не только вылечить детей с тяжёлыми врожденными пороками сердца, почек. ЦНС, опорно-двигательного аппарата, но обеспечить им приемлемое качество жизни, что возможно лишь в условиях многопрофильного детского стационара, все специалисты и службы которого имеют большой опыт в выхаживании новорождённых с этой сложной патологией. Диспансерное наблюдение за больными следует проводить до полного завершения реабилитации в течение нескольких лет.
Источник